研究TOPICS
-
2024.04.10
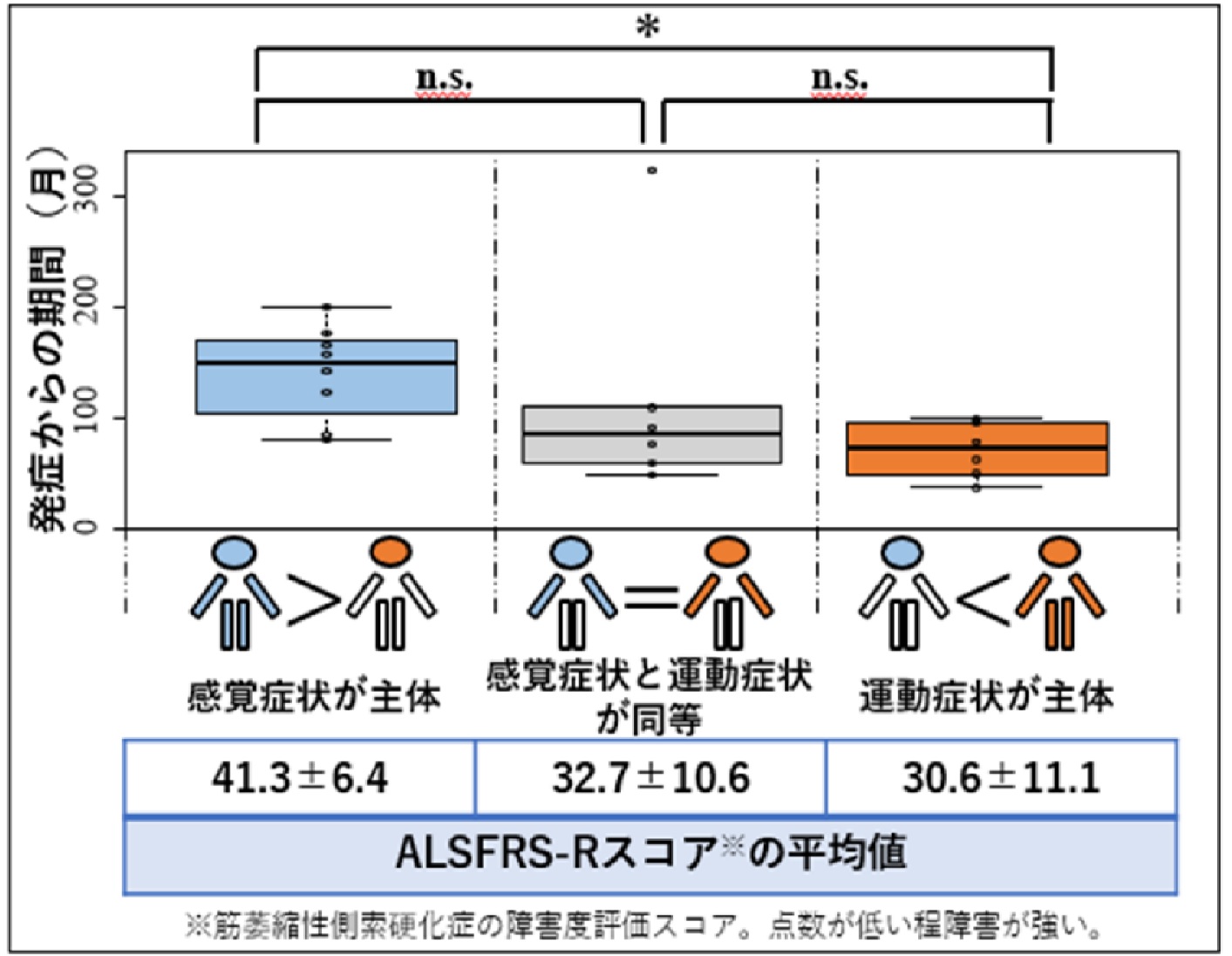
顔面発症感覚運動ニューロノパチー(FOSMN)の臨床像を解明 ~早期診断・治療・社会資源導⼊につながることが期待される~ (神経内科学分野・⼭﨑 亮准教授)
-
2024.04.03
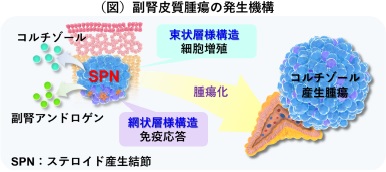
コルチゾール産⽣腫瘍の前駆病変を世界で初めて発⾒ 〜副腎腫瘍の発⽣メカニズムの解明と副腎⽪質疾患の治療への応⽤に期待〜(病態制御内科学分野 小川佳宏 主幹教授)
-

2024.03.13
人命救助した医学系学府の大学院生に対し東警察署長から感謝状贈呈
-

2024.03.06
病的ひきこもりと健康なひきこもりを区別する評価法(HiDE)開発 〜ゲーム障害・うつ病などを併存しやすい「病的ひきこもり」の早期⽀援実現へ期待~(精神病態医学分野・加藤隆弘准教授)
セミナー/学会・シンポジウム/
市民公開講座
一覧を見る
お知らせ
お知らせ一覧医学部
DEPARTMENT OF MEDICINE
各学科サイトより詳細をご確認頂けます。
大学院/大学院医学系学府
GRADUATE SCHOOL OF MEDICAL SCIENCES
各学科サイトより詳細をご確認頂けます。
